First Name Last Name
Designation Dr Lokesh Saini, MD (Pediatrics), DM (Pediatric Neurology)
Assistant Professor
Department of Pediatrics
AIIMS, Jodhpur
Case:
- A 9 month-old boy presented with global developmental delay (no head control, no cooing, no social smile),persistent stiffness of all four limbs which was non-progressive,and poor visual fixation noticed since early infancy.
- He did not have seizures, auditory inattention, regression of milestones, asymmetry of paucity of limb movements,extrapyramidal movements or history of intermittent decompensation.
- She was first-born to non-consanguineously married parents by a term, vaginal delivery with smooth perinatal transition.
- The family history was negative for any neurological disease or familial conditions.
- Anthropometric parameters were within normal limits. Head circumference was normal (43 cm).
- There were no neurocutaneous markers, facial dysmorphism or congenital malformation, skin/hair/eye changes or any signs of micronutrient deficiency.
- Neurological examination showed no social smile, no visual fixation, but an alerting response to sound was noted. Cranial nerve examination was negative for squint or bulbar involvement. He showed no head control with axial hypotonia and generalized appendicular spasticity with brisk muscle stretch reflexes and no asymmetry. Spine appeared unremarkable.
- Rest of the clinical examination was unremarkable- organomegaly was absent.
Clinical differential diagnosesof a developmentally delayed child with spasticity and no apparent perinatal insult include:
- Structural malformation (Lissencephaly, Polymicrogyria)
- Metabolic disorder (arginase deficiency; late-onset disorders of amino acid metabolism- NKH, MSUD; mitochondrial disease; organic acidemias-biotindase deficiency; cerebral folate deficiency; Sjogren Larrson syndrome)
- Genetic disease (early-onset forms of Hereditary spastic paraplegia, hypomyelinating/demyelinating leukodystrophy)
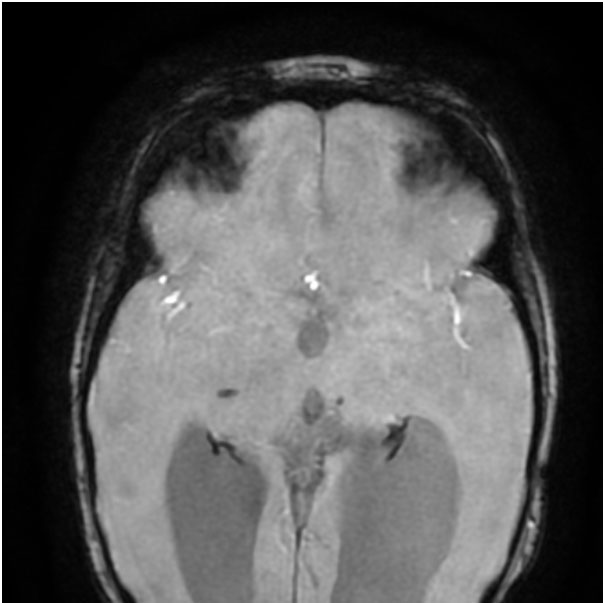
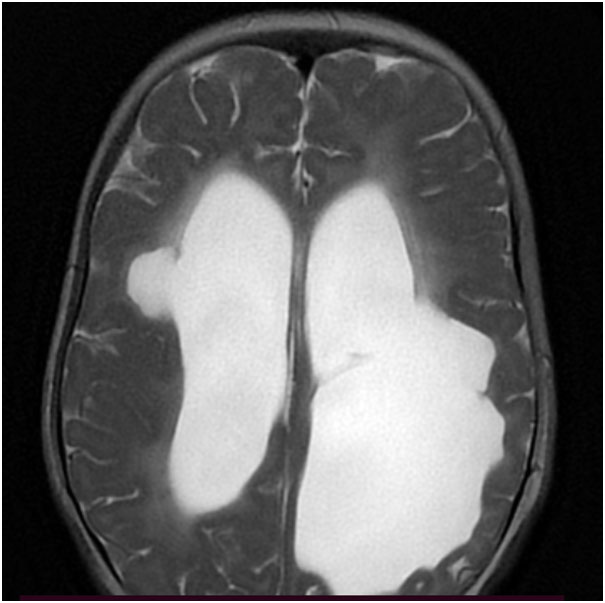
Hence, the first step in the diagnostic process is getting an MRI of the brain done. Neuroimaging in this patient (Figure 1 A-B) suggested a structural etiology (Porencephalic cysts).
- Porencephalic cysts are seen in preterm infants as a sequela to periventricular hemorrhagic infarction related to germinal matrix hemorrhage.
- Perinatal infection, perinatal arterial ischemic stroke, asphyxia, birth trauma, maternal warfarin use can result in porencephalic cysts in term as well as preterm infants.
- Porencephalic cysts may occur after antenatal or neonatal parenchymal hemorrhagic infarction in the context of other genetic conditions such asainherited coagulopathy, or thrombophilia (most often heterozygosity for factor V Leiden mutation).
- Pathogenic variants in COL4A1 result in a similar radiological picture.
In the absence of an apparent acquired etiology (uneventful pregnancy and perinatal period), inherited causes were considered. The imaging features (porencephalic cysts with microhemorrhages) pointed to a possibility of COL4A1-related disorder. Exome sequencing showed a heterozygous missense variation (pathogenic) in exon 45 of COL4A1 gene confirming the diagnosis. Parents tested negative for this variant.
COL4A1-related disorder is a genetic disorder due to a pathogenic variation in the COL4A1 gene. The COL4A1 gene encodes alpha chain of type IV collagen that forms integral part of basement membrane of cells and is responsible for the structural and functional integrity of basement membrane including that of the blood vessels. It is inherited in an autosomal dominant manner.
Presenting clinical manifestations are usually due to cerebral small-vessel disease, which include infantile hemiparesis, seizures, single or recurrent hemorrhagic strokes, ischemic stroke, and migraine later in life. These children can have systemic involvement in addition to CNS disease (outlined below).
Presence of porencephalic cysts with microhemorrhages in deep gray matter and periventricular and subcortical white matter, are the hallmark radiological features. Other imaging findings include diffuse periventricular leukoencephalopathy, lacunar infarcts, and dilated perivascular spaces.
In a child with consistent clinical features and neuroimaging features of porencephaly (without a contributing perinatal insult) with evidence of small vessel disease (microhemorrhages, infarcts) with or without a leukoencephalopathy, COL4A1-related disorder should be considered.
The spectrum of COL4A1-related disorders includes: small-vessel brain disease of varying severity in non-CNS organs which include: eye defects (retinal arterial tortuosity, Axenfeld-Rieger anomaly, cataract) and systemic findings (kidney involvement, muscle cramps, cerebral aneurysms, Raynaud phenomenon, cardiac arrhythmia, and hemolyticanemia).
- There is no curative treatment available forCOL4A1-related disorders.
- Comprehensive management of these children require the contribution of ophthalmology, nephrology and cardiology services.
- Seizures are managed with anti-seizure medications, glaucoma is managed with topical medications, symptomatic arrhythmias with anti-arrhythmic medications. Surgery is offered for cataracts and large or symptomatic intracranial aneurysms.
- Periodic brain imaging is suggested to monitor the size of aneurysm.
- Avoidance of anticoagulant exposure and activities that involve an increased risk for head trauma is important to decrease the risk for intracerebral hemorrhage.
- COL4A1-related disorders are inherited in an autosomal dominant manner.
- At least 50% of individuals diagnosed with a COL4A1-related disorder have an affected parent. The rest are denovo mutations. Since this disorder has close to 100 percent penetrance, a clinically unaffected parent is veryunlikely to carry the mutation (hence the affected child would have got the pathogenic variation denovo.)
- But since the age of clinical onset of this disorder and expressivity may be variable, it is recommended to test both parents for the mutation. In addition, careful review of parental history and revisiting their medical records, brain imaging by MRI, and ophthalmologic evaluation can help in identifying affected family members.
- In case of an apparent de novo variant, the empiric recurrence risk to siblings is approximately 1% because of the theoretic possibility of parental germline mosaicism.
T2W axial MRI brain – Bilateral periventricular white matter paucity (L>R) with porencephalic cysts and ex-vacuo dilation of bilateral lateral ventricles
SWI images – blooming along the bilateral ventricular lining indicating hemorrhagic residue